|
 |
 |
| KeyRecep |
 |
 KeyRecep is the best-suited solution for rational molecular design when the 3D structure of the target protein is unknown. Users can estimate the characteristics of the binding site of the target protein by superposing multiple active compounds in 3D space so that the physicochemical properties of the compounds match maximally with each other. (Estimation of virtual receptor model) Users can also examine relationship between chemical structures and the activities based on the multiple regression analysis with indices of conformity of each compound to the virtual receptor model and the activity values. (3D-SAR function) For compounds whose activities are unknown, users can estimate the activities based on the indices of conformity to the virtual receptor model and can perform virtual screening. (DB search function) KeyRecep rationally and strategically accelerates the molecular design projects based on hit compounds discovered by high throughput screening (HTS) or based on information on compounds from literature or patents. KeyRecep facilitates the structural expansion of such compounds to obtain lead compounds and further drug candidates. KeyRecep is the best-suited solution for rational molecular design when the 3D structure of the target protein is unknown. Users can estimate the characteristics of the binding site of the target protein by superposing multiple active compounds in 3D space so that the physicochemical properties of the compounds match maximally with each other. (Estimation of virtual receptor model) Users can also examine relationship between chemical structures and the activities based on the multiple regression analysis with indices of conformity of each compound to the virtual receptor model and the activity values. (3D-SAR function) For compounds whose activities are unknown, users can estimate the activities based on the indices of conformity to the virtual receptor model and can perform virtual screening. (DB search function) KeyRecep rationally and strategically accelerates the molecular design projects based on hit compounds discovered by high throughput screening (HTS) or based on information on compounds from literature or patents. KeyRecep facilitates the structural expansion of such compounds to obtain lead compounds and further drug candidates. |
|
|
| ADAM |
|
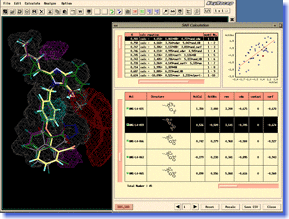
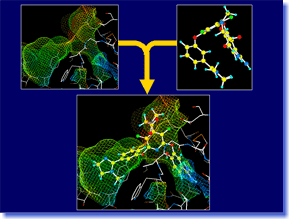 ADAM is an automated docking tool, which can predict the stable binding mode of flexible ligand molecule to target macromolecule. It is able to construct energetically favorable docking models at high speeds, exploring the conformational space of flexible ligand continuously and exhaustively. The docking accuracy and reliability in the actual drug design are the top level in the world (even in the cases of supposing induced-fit motion of target protein). Moreover, high-throughput virtual screening can be performed easily by using ADAM. In a lot of in-house projects and funded researches for drug developments, ADAM virtual screening has already achieved successes. ADAM should greatly contribute to a variety of research areas including the QSAR study and analysis of protein-ligand interaction, as well as the drug design and drug discovery. ADAM is an automated docking tool, which can predict the stable binding mode of flexible ligand molecule to target macromolecule. It is able to construct energetically favorable docking models at high speeds, exploring the conformational space of flexible ligand continuously and exhaustively. The docking accuracy and reliability in the actual drug design are the top level in the world (even in the cases of supposing induced-fit motion of target protein). Moreover, high-throughput virtual screening can be performed easily by using ADAM. In a lot of in-house projects and funded researches for drug developments, ADAM virtual screening has already achieved successes. ADAM should greatly contribute to a variety of research areas including the QSAR study and analysis of protein-ligand interaction, as well as the drug design and drug discovery. |
|
|
| LEGEND |
|
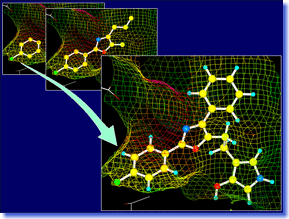 LEGEND is an efficient tool for de novo ligand design. It can automatically generate ligand candidate structures with the skeletal structures different from the known actives, based on the 3D structure of target macromolecule. The LEGEND method grows molecules by adding atoms one by one up to the specified molecular size using random numbers and force field energy calculation. This algorithm contributes to the generation of a wide variety of drug-like structures which can fit stably to the ligand-binding site. LEGEND can provide a number of innovative ideas to the medicinal chemists both in the stages of lead generation and lead development. LEGEND is an efficient tool for de novo ligand design. It can automatically generate ligand candidate structures with the skeletal structures different from the known actives, based on the 3D structure of target macromolecule. The LEGEND method grows molecules by adding atoms one by one up to the specified molecular size using random numbers and force field energy calculation. This algorithm contributes to the generation of a wide variety of drug-like structures which can fit stably to the ligand-binding site. LEGEND can provide a number of innovative ideas to the medicinal chemists both in the stages of lead generation and lead development. |
|
|
| Key3D |
|
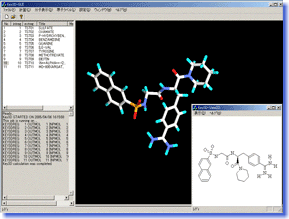 Key3D is a molecular modeling tool to convert 2D structures (chemical structural formula) of compounds drawn by ISIS-Draw or ChemDraw to highly-accurate 3D structures with additional information on atomic charge etc. It is perfectly suitable for data preparation in theoretical chemistry calculations such as force field calculation and molecular orbital calculation. Key3D is also very useful to convert a large library of commercially available or in-house compounds to a 3D database as it can process a great number of structures fully automatically. Key3D is a molecular modeling tool to convert 2D structures (chemical structural formula) of compounds drawn by ISIS-Draw or ChemDraw to highly-accurate 3D structures with additional information on atomic charge etc. It is perfectly suitable for data preparation in theoretical chemistry calculations such as force field calculation and molecular orbital calculation. Key3D is also very useful to convert a large library of commercially available or in-house compounds to a 3D database as it can process a great number of structures fully automatically.
>> For animated demonstation of Key3D, see here.
|
|
|
| Bluto |
|
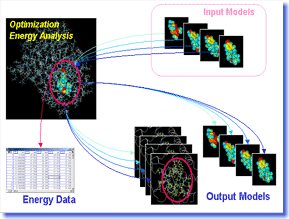 Bluto
performs energy minimization and energy analysis of
protein or protein-ligand complexes by using force field.
Users can conduct the structural optimization of docking
models of multiple ligands onto a protein continuously
and highly-accurately with easy operation, and can obtain
tabular reports of the energy analysis such as the interaction
energy. Bluto is most suitable for virtual screening
projects using the automatic docking method. Bluto
performs energy minimization and energy analysis of
protein or protein-ligand complexes by using force field.
Users can conduct the structural optimization of docking
models of multiple ligands onto a protein continuously
and highly-accurately with easy operation, and can obtain
tabular reports of the energy analysis such as the interaction
energy. Bluto is most suitable for virtual screening
projects using the automatic docking method. |
|
| Pdbfil |
|
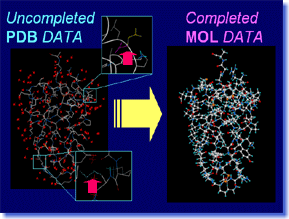 Pdbfil is a tool which automatically processes the protein coordinate data obtained from PDB, into the model in which precise molecular calculation is possible. The atoms which are missing in PDB data are appropriately compensated, after performing conformation search and energy minimization if needed. The hetero-residues and water molecules which become unnecessary for future calculation are deleted. The specified water molecules are saved, after hydrogen atoms are added and positions of them are optimized automatically. Atomic attributions like atomic charge or molecular force-field type are also assigned automatically. Pdbfil supports powerfully "one step of the beginning" of the research which uses PDB data. Pdbfil is a tool which automatically processes the protein coordinate data obtained from PDB, into the model in which precise molecular calculation is possible. The atoms which are missing in PDB data are appropriately compensated, after performing conformation search and energy minimization if needed. The hetero-residues and water molecules which become unnecessary for future calculation are deleted. The specified water molecules are saved, after hydrogen atoms are added and positions of them are optimized automatically. Atomic attributions like atomic charge or molecular force-field type are also assigned automatically. Pdbfil supports powerfully "one step of the beginning" of the research which uses PDB data. |
|
| ProSide |
|
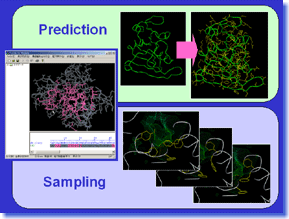 ProSide is a tool which predicts protein sidechain conformation. It is also possible to investigate the possibility of the sidechain conformation of target protein, by sampling two or more stable solutions. Since the residue-substitution by the target amino-acid sequence (read separately) is possible, ProSide can be used also for simple homology modeling, in case there are neither insertion nor deletion. Moreover, it is also possible to perform global optimization calculation of a complex, by putting ligand to a binding site, and optimizing positions and conformations of ligand and amino-acid sidechains. ProSide works out solutions to various problems related to protein sidechain conformation by high precision calculation. ProSide is a tool which predicts protein sidechain conformation. It is also possible to investigate the possibility of the sidechain conformation of target protein, by sampling two or more stable solutions. Since the residue-substitution by the target amino-acid sequence (read separately) is possible, ProSide can be used also for simple homology modeling, in case there are neither insertion nor deletion. Moreover, it is also possible to perform global optimization calculation of a complex, by putting ligand to a binding site, and optimizing positions and conformations of ligand and amino-acid sidechains. ProSide works out solutions to various problems related to protein sidechain conformation by high precision calculation. |
|
|
|
|
|